publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
2025
-
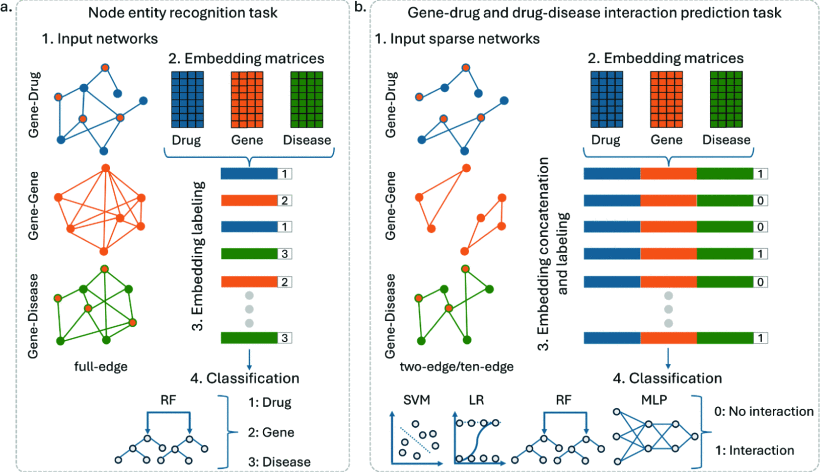 Unveiling Novel Gene-Drug-Disease Interactions with GDD-BERTLorenzo Ruggeri, Eva Viesi, Andrea Betti, and Rosalba GiugnoIn 2025 IEEE 13th International Conference on Healthcare Informatics (ICHI), 2025
Unveiling Novel Gene-Drug-Disease Interactions with GDD-BERTLorenzo Ruggeri, Eva Viesi, Andrea Betti, and Rosalba GiugnoIn 2025 IEEE 13th International Conference on Healthcare Informatics (ICHI), 2025Traditional drug discovery is a long and costly process with a low success rate. Drug repurposing (DR) offers a promising alternative by leveraging existing drugs with known safety profiles. Network-based DR models have gained attention for their ability to integrate diverse types of biological and clinical data. However, effectively combining such heterogeneous data remains a significant challenge. We propose Gene-Drug-Disease-Bidirectional Encoder Representations from Transformers (GDD-BERT), an AI model designed to identify drug-gene-disease interactions, aiding drug repositioning. It utilizes BERTwalk, a novel embedding technique based on BERT, to generate vector representations of nodes in biological networks by analyzing complex relationships through random walks. These embeddings are combined into drug-gene-disease triplets, which are used in a classifier to predict potential interactions, such as a drug targeting a protein or a protein’s link to a disease. GDD-BERT efficiently reconstructs missing interactions and excels in sparse networks, outperforming traditional methods in discovering new therapeutic applications for drugs.
-
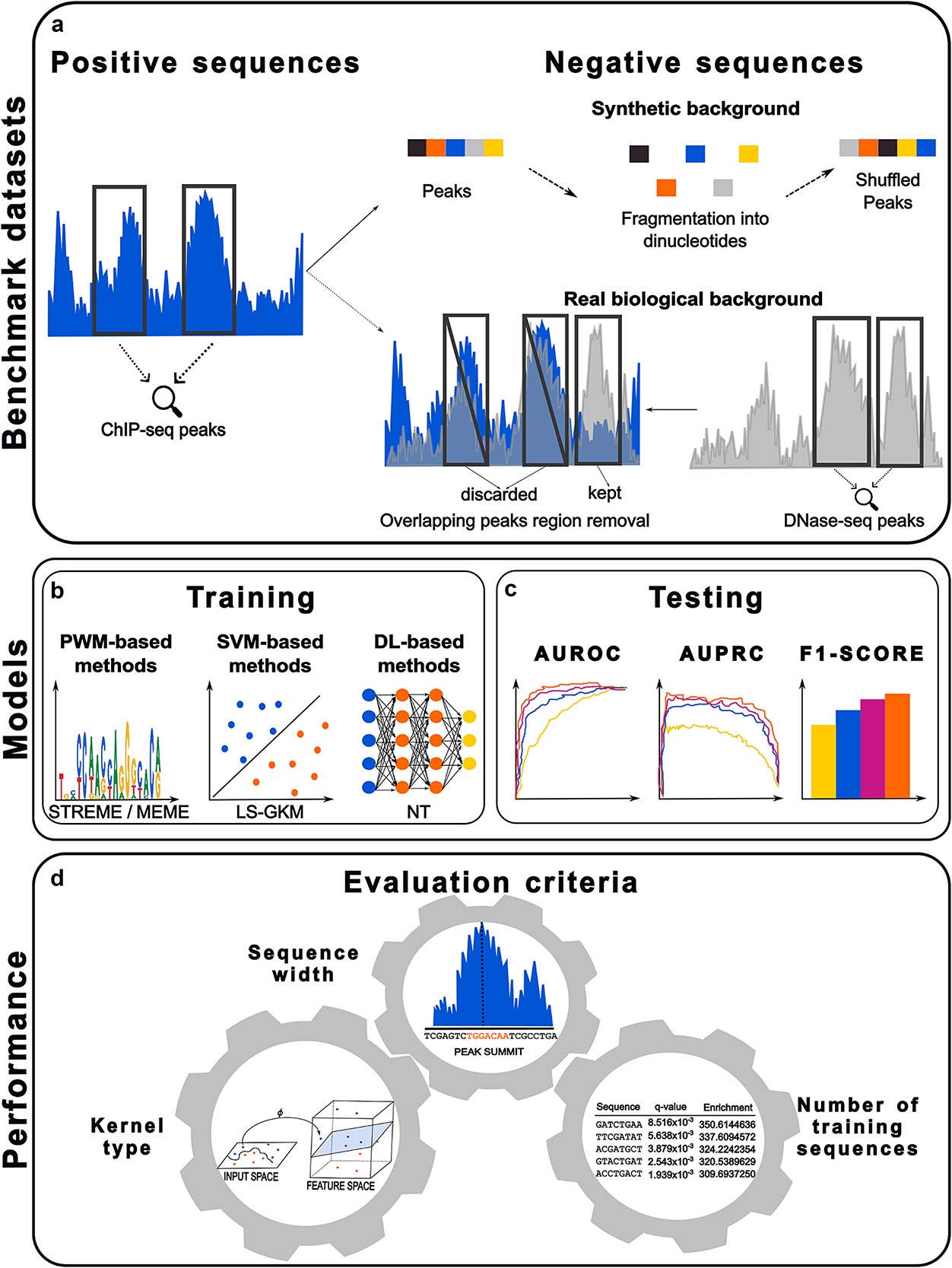 Benchmarking transcription factor binding site prediction models: a comparative analysis on synthetic and biological dataManuel Tognon, Alisa Kumbara, Andrea Betti, Lorenzo Ruggeri, and Rosalba GiugnoBriefings in Bioinformatics, Jul 2025
Benchmarking transcription factor binding site prediction models: a comparative analysis on synthetic and biological dataManuel Tognon, Alisa Kumbara, Andrea Betti, Lorenzo Ruggeri, and Rosalba GiugnoBriefings in Bioinformatics, Jul 2025Transcription factors (TFs) are essential regulatory proteins controlling the cellular transcriptional states by binding to specific DNA sequences known as transcription factor binding sites (TFBSs) or motifs. Accurate TFBS identification is crucial for unraveling regulatory mechanisms driving cellular dynamics. Over the years, various computational approaches have been developed to model TFBSs, with position weight matrices (PWMs) being one of the most widely adopted methods. PWMs provide a probabilistic framework by representing nucleotide frequencies at every position within the binding site. While effective and interpretable, PWMs face significant limitations, such as their inability to capture positional dependencies or model complex interactions. To address these, advanced methods, like support vector machine (SVM)–based, and deep learning (DL)–based models, have been introduced. Leveraging human ChIP-seq data from ENCODE, we systematically benchmarked the predictive performance of PWM, SVM-, and DL-based models across different scenarios. We evaluate the impact of key factors such as training dataset size, sequence length, and kernel functions (for SVMs) on models’ performance. Additionally, we explore the impact of synthetic versus real biological background data during model training. Our analysis highlights strengths and limitations of each approach under different conditions, providing practical guidance for selecting and tailoring models to specific biological datasets. To complement our analysis, we present a comprehensive database of pretrained SVM models for TFBS detection, trained on human ChIP-seq data from diverse cell lines and tissues. This resource aims to facilitate broader adoption of SVM-based methods in TFBS prediction and enhance their practical utility in regulatory genomics research.